Sindrome di Omenn
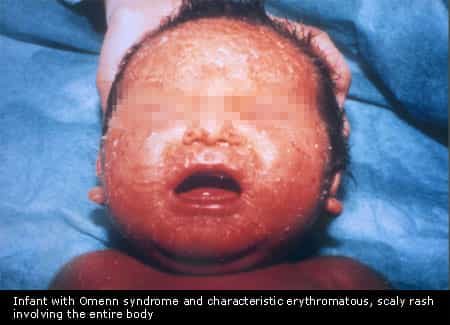
Ora per i bambini affetti dalla Sindrome di Omenn c’è una speranza in più di guarire grazie al modello animale della malattia, realizzato mediante tecniche di ingegneria genetica nei laboratori del Reparto Genoma Umano dell’Istituto di tecnologie biomediche (Itb) del Consiglio nazionale delle ricerche di Milano. Il risultato è l’ultimo prodotto del Progetto Genoma Umano che, dopo aver stabilito la sequenza di tutto il Dna dell’uomo, si dedica ora alla comprensione del suo funzionamento. «La sindrome di Omenn (SO)», spiega Anna Villa, la ricercatrice dell’Itb-Cnr che ha diretto lo studio, «è una immunodeficienza grave e appartiene al gruppo delle SCID (Severe combined immunodeficiencies), un insieme di patologie che hanno in comune un difettoso funzionamento dei linfociti B e T. Si tratta di due classi di cellule fondamentali perché sovrintendono alla risposta immunitaria che permette all’organismo di riconoscere i patogeni che hanno precedentemente infettato l’organismo, eliminandoli rapidamente e consentendo così all’individuo di far fronte a tutti gli attacchi provenienti dall’ambiente esterno. La maturazione di queste cellule è legata al buon funzionamento di numerosi geni, tra cui i geni RAG1 e RAG2, che sono alterati nella Sindrome di Omenn. Per ricreare nel topo lo stesso difetto genetico presente nei pazienti affetti da questa patologia ci siamo avvalsi di tecnologie di ricombinazione genetica. Avere a disposizione un modello animale ci consentirà di capire come nasce il difetto immunologico alla base della SO e di testare nuove terapie». Questa particolare malattia prende il suo nome da G. S. Omenn che la scoprì nel lontano 1965. La sindrime di Omenn si manifesta nei primi mesi di vita come un’immunodeficienza classica: il neonato è colpito da numerose infezioni, presenta disidratazione, malnutrizione e diarrea che ne ritardano la crescita, come avviene in tutte le SCID. A questi sintomi si accompagnano però anche fenomeni di autoimmunità : ingrossamento di fegato e milza (epatosplenomegalia), ingrossamento dei linfonodi (linfoadenopatia) ed eczema cutaneo. La malattia non trattata porta a morte certa nei primi anni di vita e l’unico intervento possibile è il trapianto di midollo con cellule di donatori istocompatibili, trapianto che deve essere effettuato precocemente, possibilmente prima della comparsa delle complicanze di tipo autoimmune. È facile comprendere quindi l’importanza del modello animale realizzato dall’Itb-Cnr, in grado senz’altro di chiarire la patogenesi dei sintomi autoimmuni e di comprendere come essi si stabiliscano. [via cnr]
Potrebbe interessarti
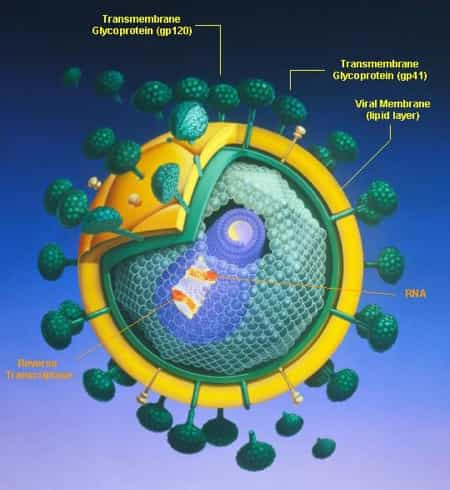
HIV: new therapeutic strategies
To eliminate HIV from the body, at a minimum, infected, quiescent T cells would need to be forced to produce viral proteins. This would cause the destruction of these cells, which would be attacked by drugs that block the spread of the virus from one cell to another. New data suggest that an intensification of… Continua a leggere HIV: new therapeutic strategies
Cellule artificiali: la nuova frontiera della terapia
Sulla nota rivista Science è stata pubblicata una ricerca destinata a rivoluzionare il futuro della medicina: la nascita della vita artificiale cioè le cellule artificiali. L’artefice di ciò è stato Craig Verter, scienziato di fama, protagonista nel 2000 del Progetto Genoma Umano, pioniere degli esperimenti di lungo corso ma anche personaggio criticato dalle commissioni etiche… Continua a leggere Cellule artificiali: la nuova frontiera della terapia

La dieta del DNA
C’è stato il tempo della frutta, della verdura, della pasta, ora è il momento della dieta del DNA. L’iniziativa è di Planet srl, che gestisce Vitalybra, un piano alimentare personalizzato ideato dal medico nutrizionista Primo Vercilli. Per provarla la nuova dieta basta sottoporsi ad un test genetico in una delle principali farmacie italiane che hanno… Continua a leggere La dieta del DNA
La variabile espressione dei geni
Dal decodificare il genoma umano ad arrivare ad una sua piena comprensione la strada è ancora molto lunga. Oggi si comincia a capire che non è solo importante l’informazione contenuta ma soprattutto la sua regolazione. Dalla regolazione, infatti, nascono le differenze tra gli individui ma anche le malattie o le predisposizioni alle malattie. Dalla collaborazione… Continua a leggere La variabile espressione dei geni